Research Progress
Recently, the research team led by Yuqian Chen at the School of Chemical Biology and Biotechnology and the School of Scientific Intelligence at Peking University, in collaboration with the National University of Singapore, developed a machine learning interatomic potential model named E2GNN based on an efficient equivariant model. Unlike traditional equivariant models that rely on complex high-order tensor representations,E2GNN encodes equivariant features using a concise scalar-vector representation and incorporates a global message-passing mechanism. This approach enables a more accurate description of long-range atomic interactions, achieving efficient and physically reasonable potential energy predictions. The related findings were published in the prestigious AI4M (AI for Materials) journalnpj Computational Materials (a top-tier journal in the Chinese Academy of Sciences classification) under the title "Efficient equivariant model for machine learning interatomic potentials," and the paper was selected as a Featured Article.

Accurately and efficiently predicting interatomic potentials is a core aspect of advancing material design and simulation. However, while existing deep learning methods have demonstrated significant potential in accelerating computations, they often struggle to balance prediction accuracy with computational efficiency, particularly in large-scale system simulations. Achieving "fast and accurate" potential predictions requires addressing two key points. Firstly, to maintain physical plausibility, the energy output of the model must be invariant to rigid-body transformations (translation and rotation), while the predicted force field must be translation-invariant and rotation-equivariant to ensure physical consistency. Secondly, while adhering to these equivariance constraints, the model design must also maintain sufficient computational efficiency to meet the demands of large-scale simulations.

Figure 1: Fundamental principles of graph neural networks and explanation of equivariant vs.invariant concepts
To address the challenges mentioned above, the research team proposed anefficient equivariant graph neural network (E2GNN) that combines both accuracy and efficiency. Unlike traditional equivariant models, which rely on complex high-order tensor representations,E2GNN adopts a concise "scalar-vector" hybrid representation to encode equivariant features and incorporates a global message-passing mechanism. This design enables the accurate capture of long-range atomic interactions, achieving high-precision and high-efficiency potential energy prediction. The synergy between global message passing and gated message-passing mechanisms further enhances the overall performance of the model.
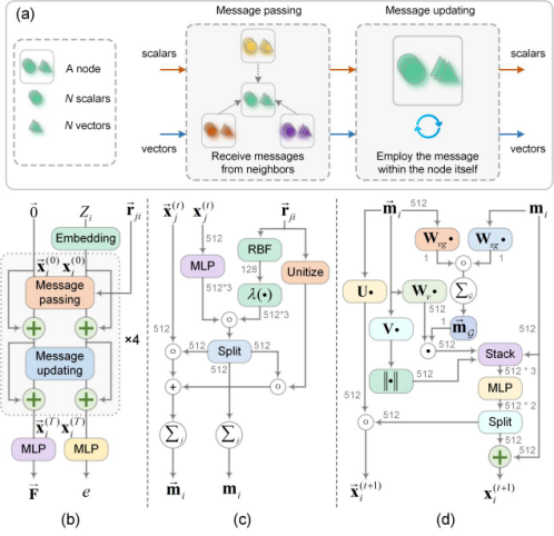
Figure 2:The overall architecture of E2GNN.
Experimental results demonstrate that E2GNN outperforms current state-of-the-art baseline models across multiple datasets, including catalysts, molecular systems, and organic isomers, while also exhibiting significant advantages in computational efficiency.Furthermore, molecular dynamics simulations of solid, liquid, and gaseous systems, conducted using the E2GNN-derived force field, achieve prediction accuracy at the level of first-principles calculations. The high-precision and high-efficiency interatomic potential prediction model proposed in this study holds great promise for accelerating the discovery of new materials, optimizing the performance of existing materials, and providing more efficient data-driven tools for material design. This advancement is poised to propel computational materials science into a new era characterized by greater precision and efficiency.
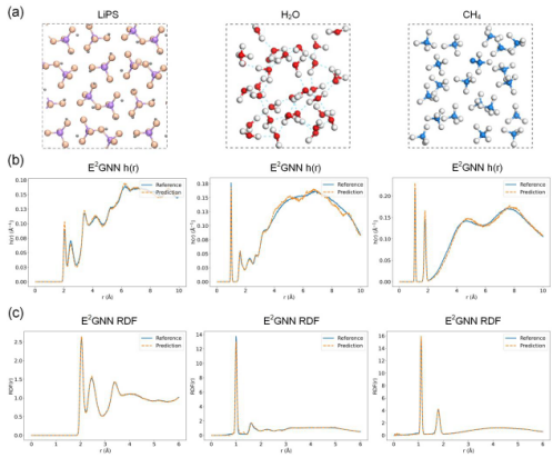
Figure 3: MD simulations for LiPS, H2O, and CH4.
Paper Link:https://www.nature.com/articles/s41524-025-01535-3





