DMOA (3,5-Dimethylorsellinic Acid)-derived meroterpenoid natural products.Nowadays, over 200 compounds of this class have been reported, characterized by their structural complexity, large quantity, diverse oxidation states, and significant biological activities. Current synthetic efforts for this class of natural products have mainly targeted compounds like simplicissin, berkeleyone A, and andrastin D. Research groups led by Porco Jr., Maimone, Newhouse, Li Houhua, and Xie Zhixiang, among others, have made contributions to this field.
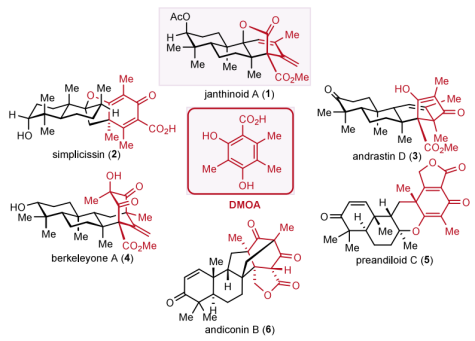
Figure 1. Naturally occurring DMOA-Derived meroterpenoids
(Image Source:J. Am. Chem. Soc.)
Janthinoid A is a natural product of this type isolated in 2021 from Penicillium janthinellum by the research group of Peng Zhang at the Yantai Tobacco Research Institute of the Chinese Academy of Agricultural Sciences. This compound exhibits in vivo inhibitory activity against non-small cell lung cancer A549 cells. Structurally, it contains six stereocenters, including four contiguous quaternary carbon stereocenters. Among these, three quaternary carbon centers are located within a highly functionalized [3.2.1] bridged bicyclic lactone structural unit.
Recently, the Zhen Yang/Zhongchao Zhang team reported the first asymmetric total synthesis of the natural product Janthinoid A. Starting from commercially available geranylacetone, the synthesis was accomplished in 14 total steps with an overall yield of 3.8%, without the need for protecting groups. The key transformations in this route include:
1)An epoxide-initiated, allene-participating cationic polyene cyclization to construct the crucial meroterpenoid synthetic intermediate, an enal.
2)An iron(III) perchlorate-mediated oxidative radical cyclization that constructs the rigid [3.2.1] bridged bicyclic lactone core skeleton bearing three contiguous quaternary carbon stereocenters in a single step.
The retrosynthetic analysis is illustrated as follows: The [3.2.1] bridged bicyclic lactone structure in 1 can be accessed via an oxidative radical cyclization from tricarbonyl substrate C. C, in turn, can be constructed from the reported D through an HWE reaction and a condensation reaction. Herein, the authors designed the use of E to build this compound via a cationic cyclization strategy. The E can be synthesized from F through a reported 1,3-acyl migration method. F itself can be prepared in three simple functional group transformations starting from commercially available geranylacetone.
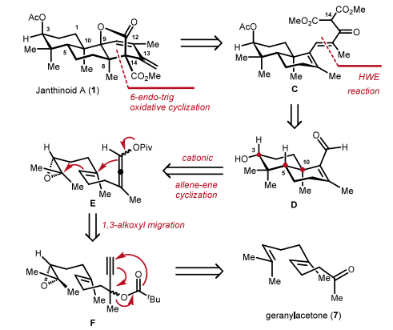
Figure 2. Retrosynthetic analysis
(Image Source:J. Am. Chem. Soc.)
The total synthesis route is as follows: First, starting from commercially available geranylacetone, the authors employed the ent-Corey–Zhang ligand to accomplish an asymmetric dihydroxylation of the substrate with 97% yield and 92% ee. Subsequently, an intramolecular S<sub>N</sub>2 reaction was used to construct an epoxide compound with an R configuration at the C3 stereocenter. Treatment with ethynylmagnesium bromide and pivaloyl chloride then afforded the propargylic ester 9. Next, using rhodium(III) trifluoroacetate, an intramolecular 1,3-acyl migration yielded the allenoate 10. Condition screening for this polyene cyclization reaction identified the optimal system asBF3·Et2O/DCM, which furnished the target product 11 in 65% yield. In a total of five steps, this crucial meroterpenoid synthetic intermediate was obtained asymmetrically.
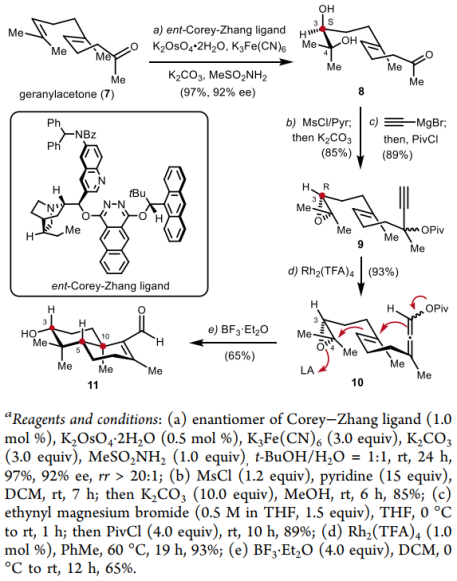
Figure 3. Synthesis ofaldehyde 11
(Image Source:J. Am. Chem. Soc.)
After obtaining this enal, the authors investigated the introduction of the side chain. First, the C3 hydroxyl group was oxidized to a ketone. Under Horner–Wadsworth–Emmons (HWE) conditions, the regioselective and purely (E)-configured enoate 14 was obtained in 69% yield over two steps. Subsequent stereoselective reduction of the ketone carbonyl with K-selectride yielded diastereomer 15 with 98% yield and a dr value of 4.8:1. Hydrolysis with lithium hydroxide then provided hydroxycarboxylic acid 16. The structures of compounds 14 and 16 were both confirmed by X-ray diffraction analysis. Esterification of the hydroxyl group gave carboxylic acid 17 in 96% yield over two steps. The carboxyl group was then converted to an acid chloride, which was condensed with dimethyl malonate to afford the tricarbonyl compound 18 in 70% yield. This set the stage for subsequent studies on the oxidative radical cyclization reaction.
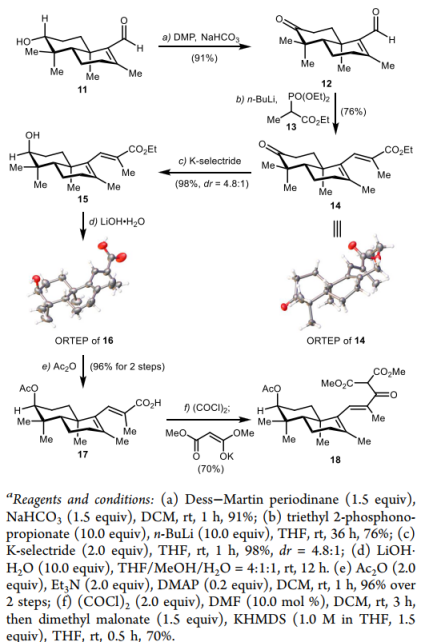
Figure 4. Synthesis ofketoester 18
(Image Source:J. Am. Chem. Soc.)
Next, inspired by Citterio's 1993 report on oxidative radical cyclization achieving E/Z isomerization of alkenes (Citterio, A. et al. Tetrahedron 1993, *49*, 7743–7760.), the authors explored conditions for the key oxidative radical cyclization. Initially applying the classic Mn(OAc)₃/AcOH system for oxidative radicalcyclization, they found only decomposition of the substrate occurred. Replacing the solvent with DCM or MeCN resulted in no reaction. When one equivalent of trifluoroacetic acidwas added to the system, 20% of the target cyclized product 19 was formed, and its structure was confirmed by X-ray diffraction. Subsequently, replacing the oxidant with ferric perchlorate (Fe(ClO₄)₃) increased the yield to 55%. Raising or lowering the reaction temperature both led to decreased yields. When the solvent was changed to DCM, the reaction halted due to the poor solubility of ferric perchlorate in this solvent. The authors also tried other Fe(III) salts such as FeCl₃ and Fe(acac)₃, none of which provided satisfactory results.Based on this, the reaction successfully constructed the congested [3.2.1] bridged bicyclic lactonecore skeleton containing three contiguous quaternary carbon stereocenters on the right side, completing the framework assembly of Janthinoid A.
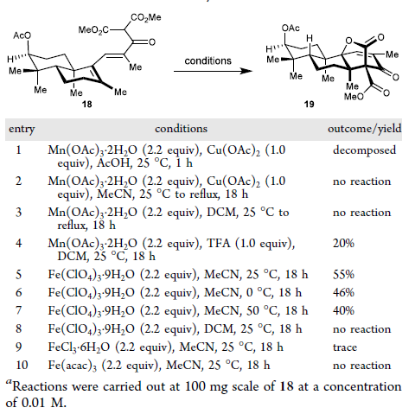
Figure 5. Condition screening for the tandem oxidative/isomerization/cyclization reaction
(Image Source:J. Am. Chem. Soc.)
In the final stage of the synthesis, only the methylenation at the C13 position of compound 19 was required to complete the total synthesis of the natural product. However, because this enone is located in a congested steric environment with abundant nearby nucleophilic acceptors, common methylenation conditions such as Wittig, Peterson, Julia, Tebbe, and Takai olefination reactions were unsuccessful. Ultimately, using LaCl₃·(LiCl)₂ as an additive, methyl lithium addition followed by Burgess dehydration achieved this methylenation step in 42% yield, accomplishing the first asymmetric total synthesis of the natural product Janthinoid A.
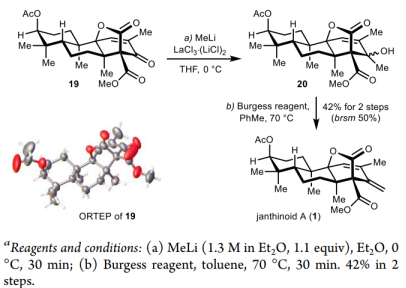
Figure 6. Asymmetric synthesis of Janthinoid A (1)
(Image Source:J. Am. Chem. Soc.)
Finally, to elucidate the mechanism of the isomerization/oxidative cyclization reaction, the authors conducted computational chemistry studies at the PWPB95-D3/def2-QZVPP//ωB97X-D/def-TZVPlevel of theory. All calculations were performed under acetonitrile solvation conditions, with the relevant results shown in the figure below.
Regarding the E/Z isomerization process, the optimal pathway identified through screening involves the reversible 5-exo-trig radical addition of the ester carbonyl oxygen atom to the alkene. This step exhibits a relatively high reaction rate, with the transition state TS1 having an activation Gibbs free energy of 13.4 kcal/mol relative to the radical intermediate Int1a. From a stereoselectivity perspective, the convex-side addition (TS1) is slightly more favorable than concave-side addition (TS1'). This is followed by a rapid C–C single bond rotation (TS2). The C–O bond dissociation step has an activation Gibbs free energy of 10.2 kcal/mol (transition state TS3), ultimately leading to the formation of intermediate Int4a.
A conformational equilibrium exists between Int4a and Int4b. Int4a, with its coplanar ketone carbonyl and ester group conformation, is less stable, and its spin density is primarily distributed on the ketone carbonyl oxygen atom and the diene moiety. In contrast, in Int4b, the O=C(Me)–CO₂Me dihedral angle is close to 90°, and the spin density is concentrated at the α-C position of the malonate ester group. After the conformational change, Int4b readily undergoes a 6-endo-trig cyclization to form the stable intermediate Int5.
Other less competitive pathways include an E/Z isomerization process based on 3-exo-trig cyclization, which has a calculated effective activation energy of 28.8 kcal/mol, and a 4-endo-trig radical addition process requiring an activation energy of 25.3 kcal/mol. Both these pathways are energetically unfavorable under the reaction conditions.
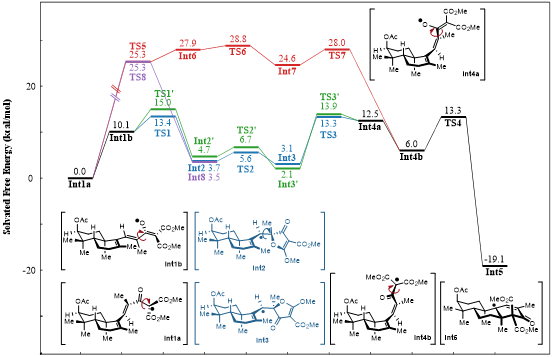
Figure 7. DFT calculation results
(Image Source:J. Am. Chem. Soc.)
Summary
The team led by Zhen Yang and Zhongchao Zhang achieved the first asymmetric total synthesis of the meroterpenoid natural product Janthinoid A, starting from commercially available geranylacetone. The synthesis was accomplished in a protecting-group-free sequence of 14 total steps. Key strategic innovations include:
1)An epoxide-initiated, allene-participating cationic polyene cyclization to construct the crucial enal intermediate.
2)An iron(III) perchlorate-mediated oxidative radical cyclization that forged the characteristic [3.2.1] bridged bicyclic lactone core on the right side of Janthinoid A.
This work provides a new synthetic pathway for the study of other natural products possessing similar structural frameworks.
The PhD candidate Fu Tang and Associate Research Fellow Zhongchao Zhang of Peking University are co-first authors of this work. PhD candidate Zhilin Song and Dr. Yuanhe Li made significant contributions to the DFT computational studies. Professor Zhen Yang and Associate Research Fellow Zhongchao Zhang of Peking University are co-corresponding authors. This research was supported by the National Natural Science Foundation of China, the Guangdong Provincial Natural Science Foundation, the Shenzhen Basic Research Program, the Shenzhen-Hong Kong Institute of Brain Science Innovation, the Shenzhen Bay Laboratory, and the Shenzhen Outstanding Talents Training Fund.
全文链接:https://pubs.acs.org/doi/10.1021/jacs.4c17480?articleRef=test
Asymmetric Total Synthesis of Janthinoid A
Fu Tang, Zhong-Chao Zhang,*Zhi-Lin Song, Yuan-He Li, Zi-Hao Zhou, Jia-Jun Chen, Zhen Yang.*





